DNA-Metabarcoding
In der Biodiversitätsforschung ist die Bestimmung von Arten in einer Region oder einem Habitat essenziell. Klassischerweise werden diese Erhebungen über Beobachtungen durchgeführt, d.h. es werden z.B. die Vögel einer Region mit Fernglas, Fische über Netz- oder Elektrobefischung oder Käfer mit Hilfe von Bodenfallen erfasst. Die Bestimmung setzt eine umfassende taxonomische Expertise voraus. Die Bestimmung ganzer Artgemeinschaften aus Mischproben (z.B. Malaisefallen, Bodenfallen) oder direkt aus Umweltproben (z.B. Wasser, Sediment, Luft) über abgegebene Umwelt-DNA (engl. Environmental DNA, eDNA) kann genetisch über das Verfahren „DNA-Metabarcoding“ durchgeführt werden. Ähnlich wie in der Forensik werden DNA-Moleküle für die Zuweisung einer Identität genutzt.
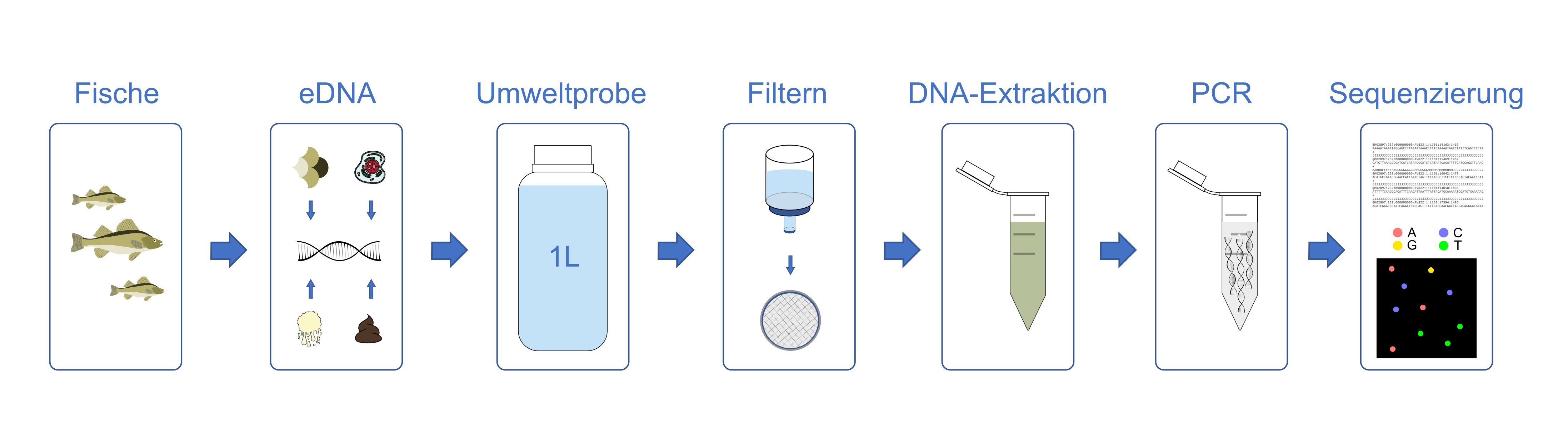
Die DNA wird entweder aus der Mischprobe gewonnen, z.B. durch Homogenisierung der Organismen, oder direkt aus der Umweltprobe extrahiert, z.B. durch Filtration des Wassers (Abb. 1). Ist die DNA extrahiert, so wird eine Polymerasekettenreaktion (PCR) durchgeführt, in der ein charakteristisches Markergen (DNA-Barcode) amplifiziert wird. Zur Amplifikation werden DNA-Sonden, sogenannte Primer, eingesetzt, die möglichst den DNA-Barcode aller in der Probe befindlichen Zielorganismen amplifiziert. Die DNA-Fragmente, auch Amplicons genannt, werden auf einem Hochdurchsatzsequenzierer ausgelesen. In der Regel werden je Probe viele Tausend bis zu Millionen Sequenzen generiert, um die Artgemeinschaft möglichst vollständig zu erfassen.
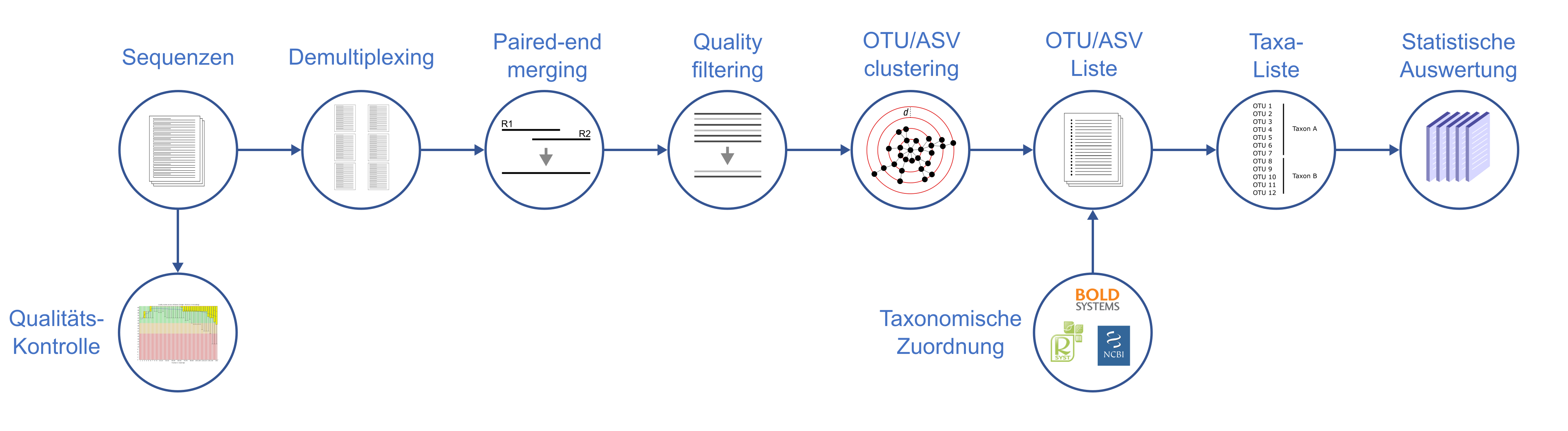
Liegen die Sequenzen vor, so erfolgt der bioinformatische Teil der Analyse (Abb. 2). Nach einer technischen Kontrolle erfolgt die Aufteilung nach verschiedenen Proben anhand von eingesetzten Markierungen (sog. Indexe). Die von vorne und hinten gelesenen Sequenzen werden zusammengefügt (paired-end merging), überflüssige Bereiche sowie Bereiche schlechter Quailtät werden entfernt und anschließend die Sequenzen nach Ähnlichkeit zusammengefasst (OTU clustering). Diese Sequenzliste wird nun genommen und gegen eine Barcode-Referenzdatenbank verglichen (z.B. BOLD). Über den Vergleich werden den Sequenzen taxonomische Namen zugeordnet und mit der finalen Liste können Analysen durchgeführt werden.
Wichtig ist, dass es unterschiedlichste Methoden an allen Schritten der Analysen gibt. Insbesondere die Wahl der Primer als auch die bioinformatischen Methoden haben einen großen Einfluss auf die Wahl der Ergebnisse und müssen daher sorgfältig geprüft werden.