DNA Metabarcoding
Identifying the species in a region or habitat is essential to biodiversity research. Traditionally, these surveys are carried out by direct observation, e.g. birds are observed using binoculars, fish using net or electro-fishing, or beetles using pitfall traps. The identification of species requires comprehensive taxonomic expertise. However, this task has been made much easier in recent years by the advent of DNA metabarcoding, which allows entire biological communities to be identified from mixed samples (e.g. malaise traps, pitfall traps) or environmental samples (e.g. water, sediment, air) based on environmental DNA (eDNA). Similarly to forensics, DNA molecules are used to assign the identity of species.
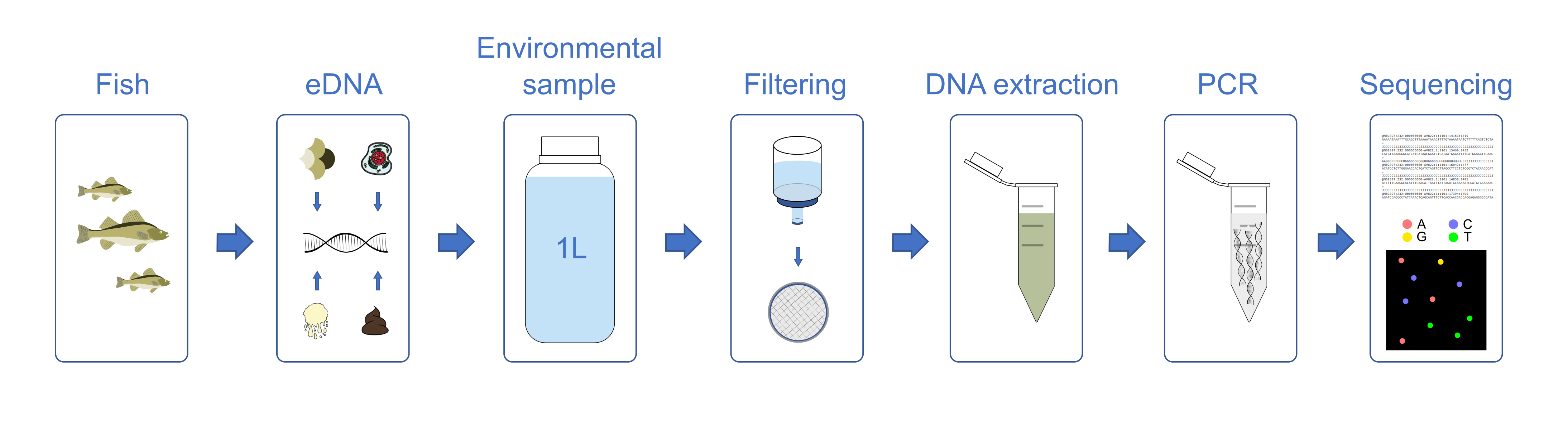
DNA is either obtained from a mixed sample, e.g. by homogenizing the organisms, or extracted directly from an environmental sample, e.g. by filtration of water (Fig. 1). Once the DNA has been extracted, a polymerase chain reaction (PCR) is carried out in which a characteristic marker gene (DNA barcode) is amplified. DNA probes, so-called primers, are used for amplification of the DNA barcode of all target organisms in the sample. The resulting DNA fragments, also called amplicons, are read out on a high-throughput sequencer. As a rule, many thousands to millions of sequences are generated for each sample in order to record the species composition as completely as possible.
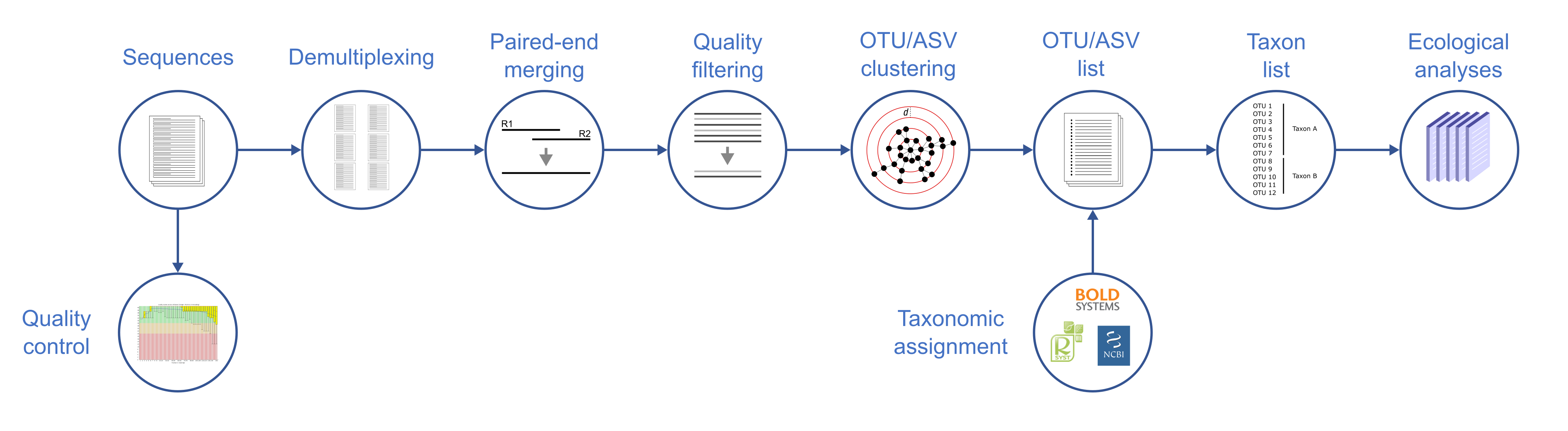
Once the sequences are available, the next step is bioinformatic analysis (Fig. 2). After a technical check, the sequences are assigned to their respective samples based on unique identifiers (“indices”). The sequences read from the forward and reverse directions are combined (paired-end merging), superfluous and low-quality areas are removed, and then the sequences are grouped according to similarity (OTU clustering). The resultant sequence list is then compared against a barcode reference database (e.g. BOLD) to assign taxonomic names, after which analyses can be carried out with the final list.
It is important to note that there are different possible approaches at each step of the analysis. In particular, the choice of primers and bioinformatic methods can have a major influence on the results, and therefore requires careful attention.